This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.

This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.
Scientific Director of the Research Institute of Fox Chase Cancer Center
Professor










T lymphocyte development and transformation
The Wiest lab employs genome wide approaches to gain fundamental insights into the molecular control of the development of T lymphocytes and other hematopoietic cells and then exploits that information to improve the efficacy of cancer treatment. The lab focuses on four main areas. Project 1 seeks to identify the causal mutations responsible for previously unsolved immunodeficiency cases, in collaboration with investigators at the University of California at San Francisco (Figure 1). Project 2 intends to establish a three-dimensional model of the genome restructuring responsible for specification of γδ lineage T cells, and then to exploit those insights to edit the genomes of γδ lineage T cells a maximize their potency as anti-cancer immune effectors (Figures 2 and 3). Project 3 focuses on how an understudied class of molecular effectors, ribosomal proteins, control normal hematopoiesis and the potential of hematopoietic progenitors for transformation (Figures 4 and 5). The goal of Project 4 is to devise novel ways to target the Ras/MAPK pathway, which is activated in 85% of human cancers. They have determined that the ERK2 substrate interaction domains have opposing roles in cancer progression and wish to exploit this insight therapeutically (Figure 6).
Molecular control of T lymphocyte development and function and malignant hematopoiesis
Project 1: Previously, research into the molecular causes of inherited defects in the human human system, so called immunodeficiencies including servere combined immunodeficiency (SCID), have led not only to a better understanding of human immune cell development, but has also led to the discovery of novel therapeutic targets for cancer treatment (e.g., BTX and Ibrutinib). Accordingly, we combine exome and whole genome sequence analysis with screening of candidate genes in zebrafish to identify the gene mutations causing SCID (Figure 1). Using this approach, we have already identified three novel causes of immunodeficiency (STN1, ARPC1B, and BCL11B) and are currently analyzing two more novel immunodeficiency genes. The role of these genes in the etiology, diagnosis or treatment of hematologic malignancies is also being explored.
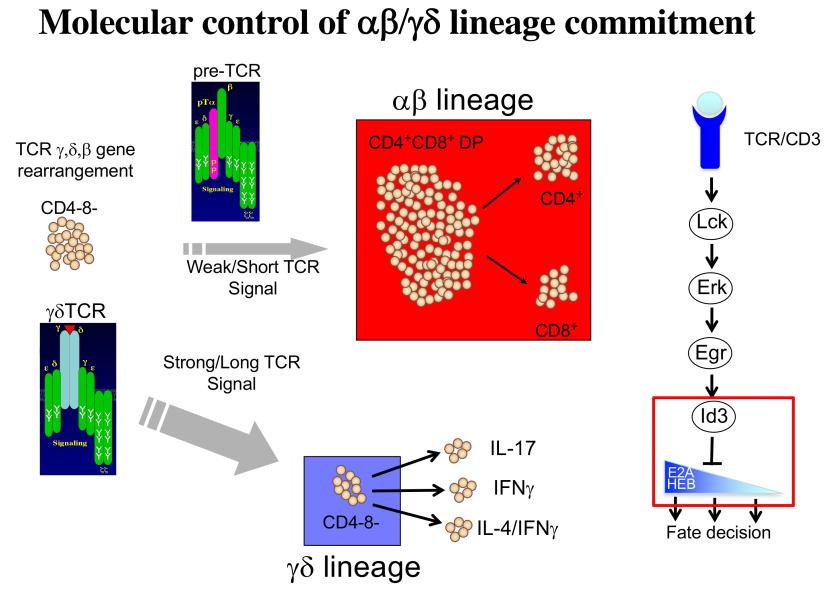
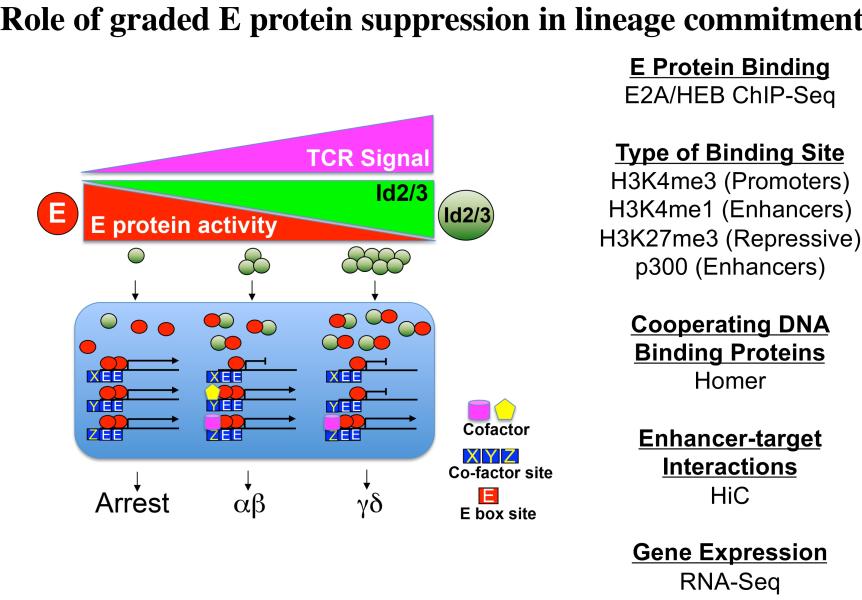
Project 2: T lymphocytes recognize and destroy invading pathogens through an assembly of proteins called the T cell antigen receptor (TCR) complex. The TCR has protein subunits that are highly variable and responsible for target recognition (either αβ or γδ) and subunits that are invariant proteins and serve to transmit signals (CD3γδε and ζ). This critical protein assembly (the TCR) controls not only the behavior of mature T lymphocytes but also their development in the thymus. My laboratory seeks to understand how T cell development is controlled by the TCR and was the first to demonstrate that this complex plays a critical role in specification of the two majory T lineages, αβ and γδ, which arise from a common immature precursor in the thymus. Importantly, γδ T cells are a long overlooked immune cell population that is increasingly understood to be critically important in immune responses to cancer. We have found that the nature of the signal transduced by the TCR plays a key role in not just specifying the γδ fate, but also influencing the functions they perform. Moreover, we have identified the signaling axis involved, which exerts its effects through the graded reduction of the activity of E box DNA binding proteins (E proteins), which are critical regulators of lymphocyte development (Figure 2). We are currently building a global three-dimensional genomic regulatory network assembled around E protein targets that are differentially occupied during fate specification (Figure 3). Our ultimate goal is the use this information in conjunction with genome editing to produce optimized anti-cancer γδ T cell effectors to deploy in adoptive cellular therapy.
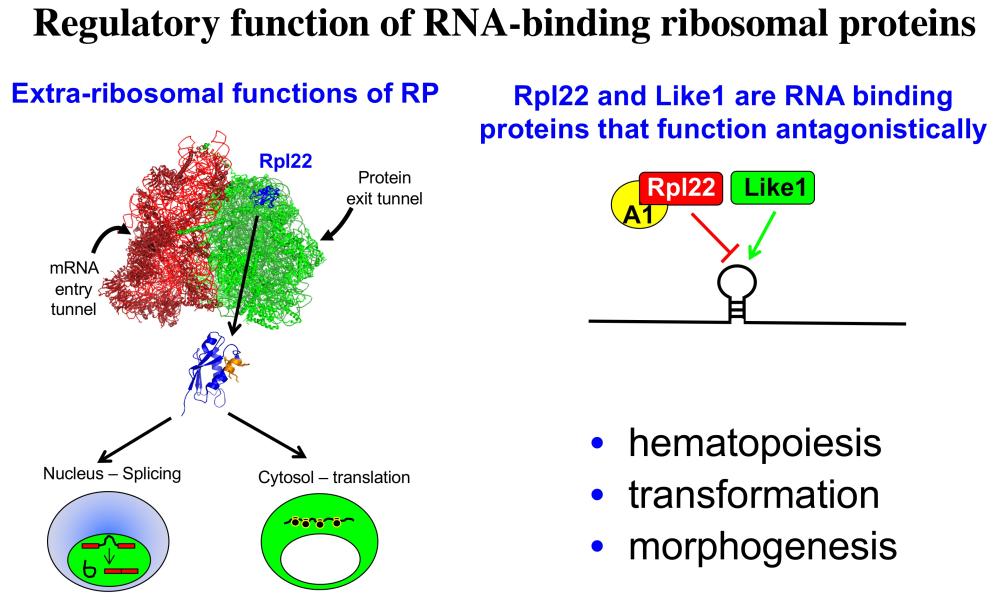
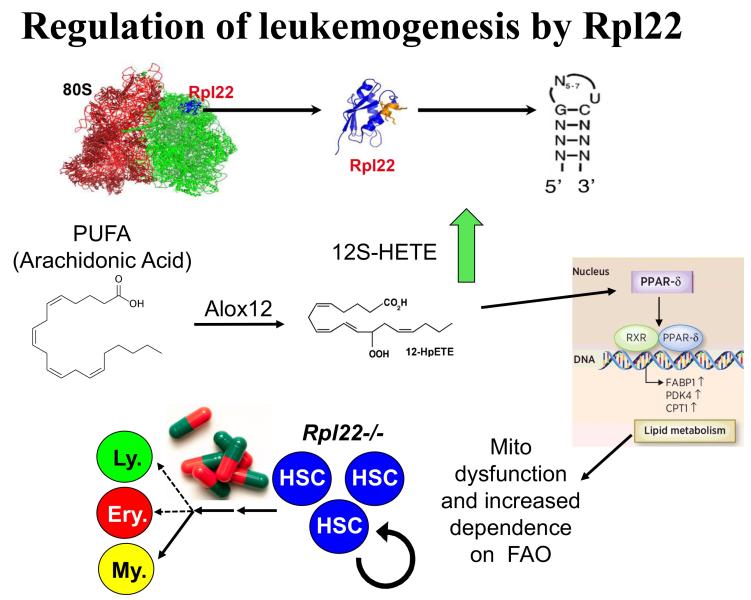
Project 3: Our efforts to understand the molecular basis for αβ/γδ T lineage commitment, led us to identify an unusual molecular effector that plays a critical role in this process, the ribosomal protein, Rpl22. Ribosomal proteins (RP) have historically been viewed as supporting the ribosome’s ability to synthesize proteins; however, emerging evidence suggests that RP, many of which are RNA-binding proteins, can actually play critical regulatory roles that control both normal development and transformation. We have found this to be true for Rpl22. Its function is selectively required for development of certain hematopoietic cells including αβ lineage T cells. Importantly, Rpl22 also regulates the emergence and behavior of hematopoietic stem cells, particularly their potential for transformation. Of note, the function of Rpl22 as a tumor suppressor is antagonized by its highly homologous (73% identical) ribosomal protein paralog, Rpl22-Like1 (Like1), which promotes development and transformation. Accordingly, developmental outcomes and risk for transformation are controlled by the antagonistic balance of Rpl22 to Like1 (Figure 4). These proteins do not appear to regulate development by altering the function of the ribosome, but instead appear to do so by functioning away from the ribosome, by binding particular RNA targets and controlling either their splicing or their translation. Efforts are ongoing to identify the collection of cellular targets through which they function as well as the molecular basis by which such similar proteins as Rpl22 and Like1 exert opposing functions. We have identified one such target, the mRNA encoding the lipoxygenase Alox12, which controls the transformation potential of hematopoietic progenitors by controlling their metabolic dependence on fatty acid oxidation (FAO), a process on which stem cells and cancer stem cells typically depend (Figure 5). These studies should inform the etiology of a number of human cancers including acute lymphoblastic leukemia (ALL), myelodysplastic syndromes (MDS), and acute myelogenous leukemia (AML), and will likely reveal new therapeutic entry points in these difficult diseases.
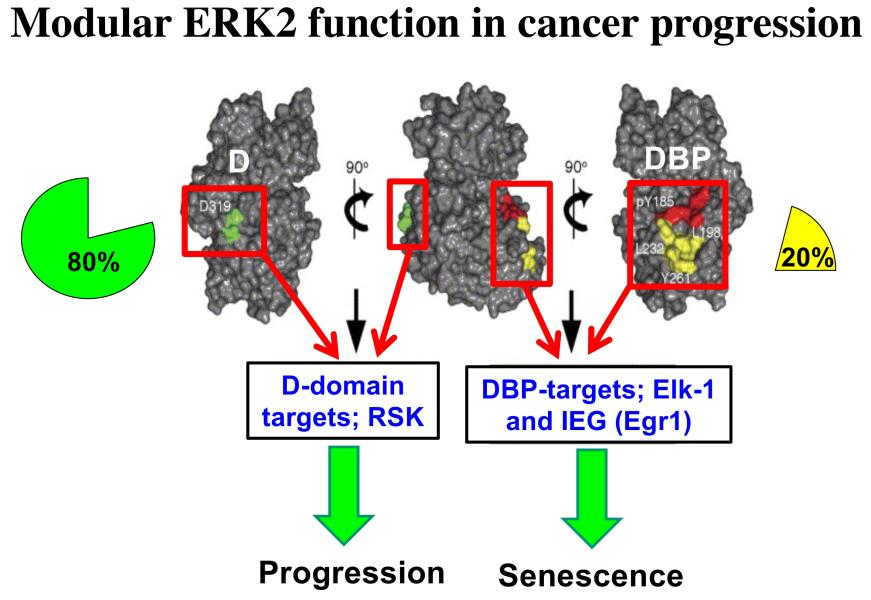
Project 4: The Ras/MAPK pathway is activated in ~85% of human cancers, but to date efforts to target this pathway therapeutically by focusing on the active sites of pathway enzymes have had limited success. We seek to develop effective therapeutic interventions for this pathway by taking a different approach, targeting the substrate interaction domains of ERK proteins, specifically ERK2. ERK2 has two different substrate interaction domains, the D domain and the DEF-binding pocket (DBP). Interestingly, we have found that the D and DBP domains of ERK2 have opposing roles in cancer progression, with D supporting progression and DBP antagonizing this process by promoting senescence (Figure 6). Consequently, we seek to understand the molecular basis for the functioning of these domains and how we can block the function of the D domain therapeutically to attenuate cancer progression.
OHSU-led effort results in largest cancer dataset of its kind
OHSU Knight Cancer Institute Press Release (Oct 17, 2018)
This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.