

This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.

This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.
Professor
Co-Leader, Cancer Signaling and Microenvironment Program
Director, Organic Synthesis Facility
Director, Molecular Modeling Facility
Adjunct Professor, University of Pennsylvania School of Medicine
Adjunct Associate Professor, Drexel University College of Medicine
The Dunbrack group concentrates on research in computational structural biology, including homology modeling, fold recognition, molecular dynamics simulations, statistical analysis of the PDB, and bioinformatics. In developing these methods, we use methods from various areas of mathematics and computer science, including Bayesian statistics and computational geometry. We place an emphasis on large-scale benchmarking of new methods and comparison with existing methods. We are interested in applying comparative modeling to important problems in various areas of biology. Areas of particular interest include DNA repair, proteases and other peptide-binding protein families, and membrane proteins.
Our software development uses visual programming environments, such as Visual Studio, and modern programming languages such as object-oriented C++ and C#. Our goal is to produce easy-to-use, professional software for use in our own research as well as to be distributed to other research groups around the world.
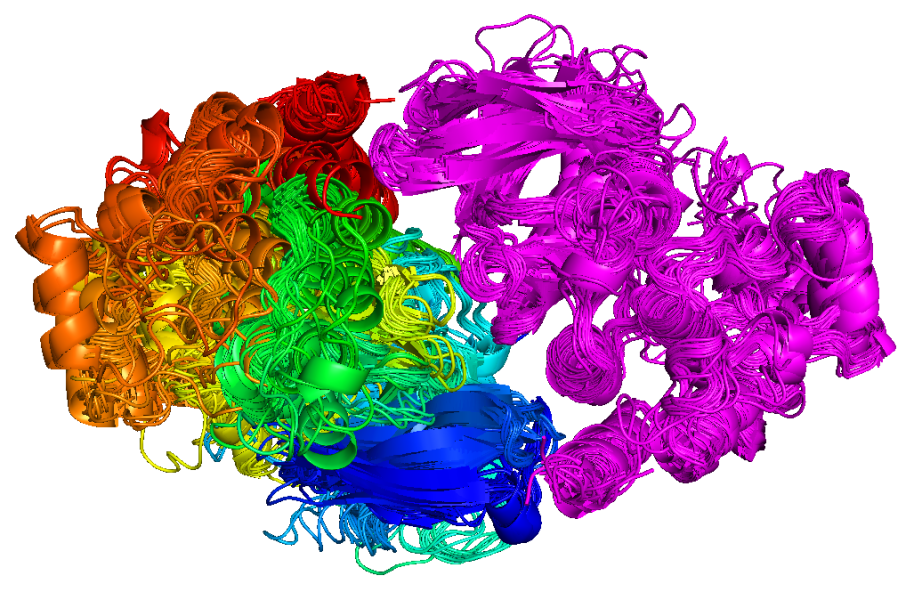
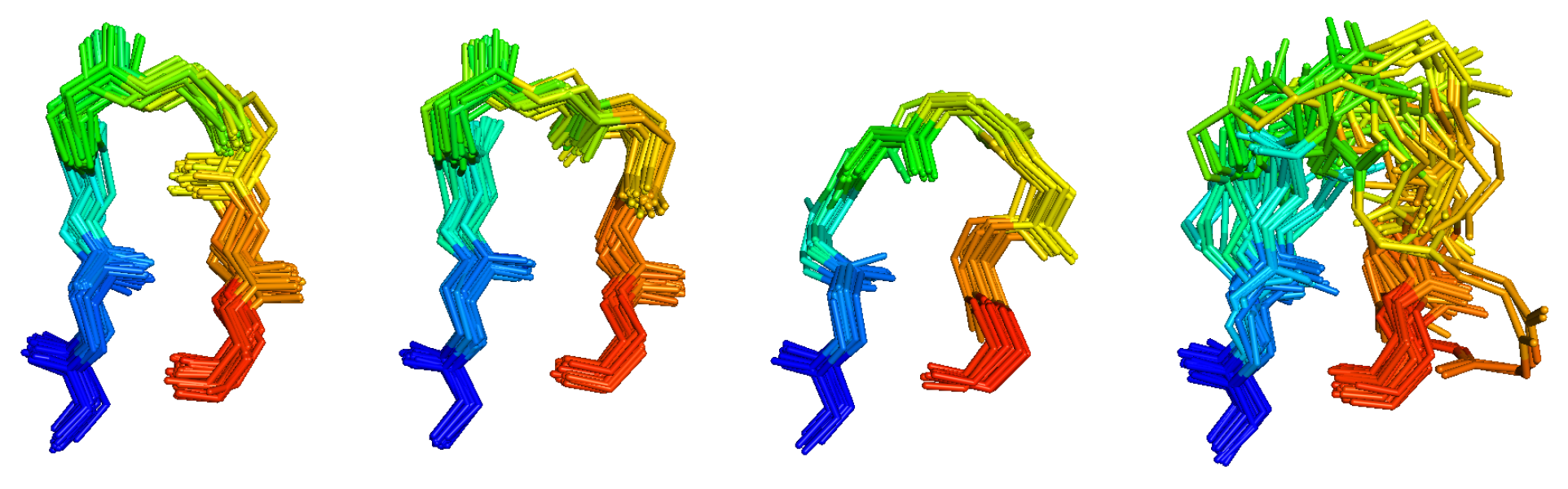
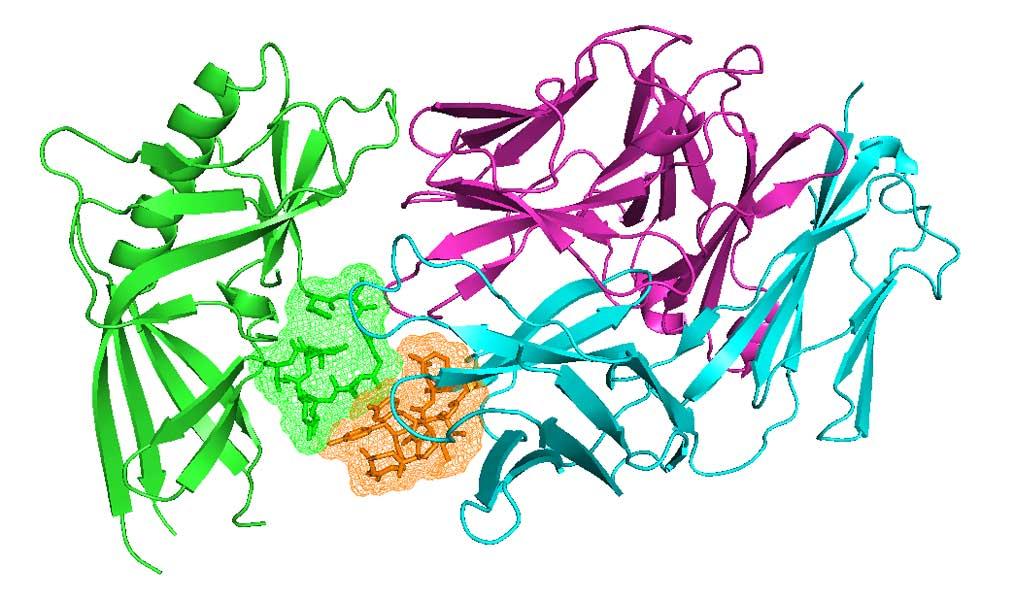
The research in my group is concentrated on statistical structural bioinformatics, the development of methods for structure prediction of proteins and protein complexes, and the application of these methods to problems in cancer biology. Our statistical studies include Bayesian and non-parametric statistical analyses of main-chain and side-chain bond angles and dihedral angles used in methods for protein structure prediction and structure determination and validation. We also analyze the structures in particular protein families, such as kinases and antibodies, across the whole structure database to develop insights that will lead to better design of cancer therapeutics. We have performed clustering and statistical analysis of protein-protein interactions in crystals as a means of determining accurate biological assemblies for each PDB entry and developing hypotheses on biologically relevant interactions that may have been overlooked by authors of structures. Our structure prediction methods include recently developed software for predicting the structures of protein complexes including both homo- and heterooligomers based on the biological assemblies of templates in the Protein Data Bank. I believe strongly in making statistical analyses, databases, and software available to all non-profit research groups, which we do via our website, http://dunbrack.fccc.edu.
I am very fortunate to work in one of the country’s leading cancer research centers and cancer hospitals, allowing me to extend and apply my interests in structural biology to problems in cancer biology. Through the Molecular Modeling Facility at Fox Chase, which I direct, we have worked with nearly all of my colleagues who work in every area of cellular or molecular biology, using structure prediction to help them explain or design experiments in any system of interest. With the advent of germline and tumor gene sequencing in cancer patients, I believe there is an opportunity to apply knowledge gained from structural bioinformatics and structure prediction in guiding the design of therapeutics in precision medicine.
*=joint corresponding authors




We have several ongoing projects in structural bioinformatics, whose purpose is twofold: understanding the determinants of the structures and dynamics of proteins and protein complexes; and providing information that is useful for improving structure prediction.
We have developed several programs for protein structure prediction, and made these publicly available via our website.
This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.










