


This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.

This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.
Amino Acid Genetics and Human Disease
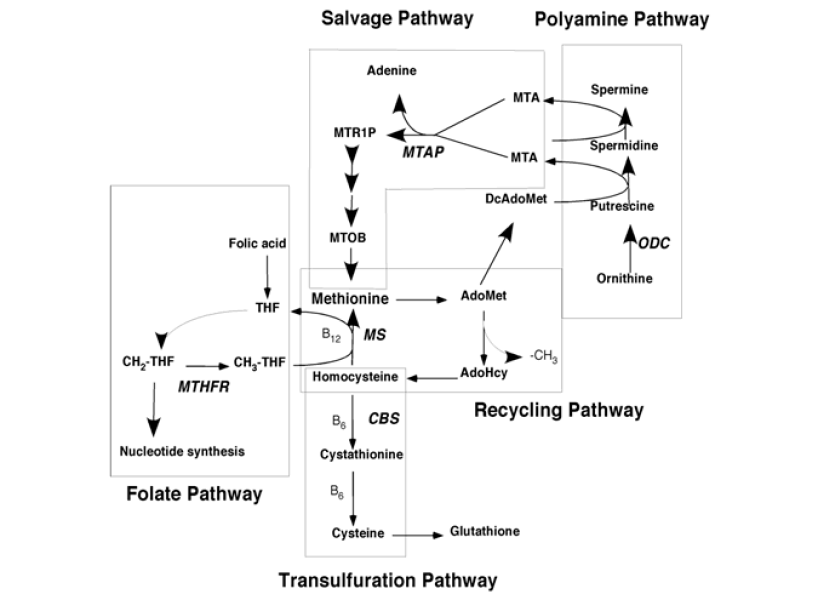
Amino acids are the building blocks of proteins and other important biological molecules. The sulfur containing amino acid methionine is especially interesting as alterations in methionine metabolism and methionine-related pathways are found in many human diseases, including rare inborn errors of metabolism to common diseases such as cardiovascular disease and cancer. Methionine metabolism is a central metabolic node connecting a variety of important metabolic processes including folate/nucleotide biosynthesis, sulfur and redox metabolism, choline and lipid metabolism, polyamine metabolism, and a wide slew of methylation reactions involving the key methyl donor S-adenosylmethionine. The lab’s interest is understanding how mutations that affect key enzymes in methionine metabolism cause disease. Currently there are four major projects in the lab. Two concern the methionine salvage pathway gene, methylthioadenosine phosphorylase (MTAP). A third project involves studying a rare inborn error of methionine metabolism (CBS deficiency) using mouse models. The final project is related to understanding how alterations in amino acid metabolism contributes to renal cell carcinoma.
There are four major areas of research in the lab. Below we will describe each of the major projects.
Methylthioadenosine phosphorylase (MTAP) is a key enzyme in the methionine salvage pathway whose function is to convert 5¢-deoxy-5¢-methylthioadenosine (MTA) into methionine. Inactivation of MTAP, often by homozygous deletion, is found in both solid and hematologic malignancies and is one of the most frequently observed genetic alterations in human cancer. Previous work from our lab and others has established that MTAP can act as a tumor suppressor gene, affecting cellular functions related to migration and invasion. However the precise mechanism by which MTAP loss promotes tumorigenesis is still unclear. One possible mechanism involves the accumulation of MTA, which is secreted by MTAP-deleted tumor cells. Structurally, MTA closely resembles adenosine and evidence indicates that it can interact with adenosine receptors. Large-scale genetic screens using shRNA have established that MTAP-deleted cells are especially sensitive to knockdown of a specific protein arginine methyltransferase enzyme (PRMT5), which is responsible for the post-translational symmetric dimethylation modification of arginine residues (sDMA). This modification is frequently observed in proteins involved in mRNA maturation. Our lab has shown that loss of MTAP or addition of extracellular MTA causes a dramatic reduction of the steady-state levels of sDMA-containing proteins. Significantly, when extracellular MTA is added, no increase in intracellular MTA occurs, suggesting that the reduction in sDMA-ylation is due to a signal transduction process. However, our data also suggest that enzymatically inactive MTAP protein itself, independent of MTA, can affect mRNA levels and antagonize the effects of MTA. We hypothesis MTAP mediates its tumor suppressor function via two different mechanisms: one involving MTA as an oncometabolite and the other a direct role for the MTAP protein. To test this hypothesis we are identifying specific proteins and arginine residues that are differentially methylated in response of loss of MTAP using state of the art LC-MS/MS proteomic technologies. We are also trying to determine the mechanism by which extracellular MTA alters protein sDMA-ylation. One specific hypothesis we are testing is that sDMA-ylation is being altered via MTA’s effect on membrane bound adenosine receptors. Another focus of our work is to try and clarify the enzymatic and non-enzymatic roles of MTAP. These studies make use a pharmacologic inhibitor of MTAP that inhibits its enzyme activity, but does not affect MTAP protein levels. These studies are significant because they provide a potential mechanism for understanding the role that MTAP deletion plays in tumorigenesis and may lead to novel therapeutic strategies for MTAP-deleted tumors. In addition, these studies will shed light on how a “housekeeping” metabolic enzyme can have a novel role as a tumor suppressor gene.
Based on genetic data, we estimate that there are approximately 260,000 new MTAP-deficient cancers diagnosed in the United States every year. Since MTAP protein is found at high levels in all normal tissues, a therapy that could specifically target MTAP-deficient cancer cells has the potential to be very effective and tumor specific. The normal enzymatic function of MTAP is to convert the polyamine biosynthetic byproduct 5’deoxy-5’-methylthioadenosine (MTA) to the purine base adenine and the sugar, 5-methylthioribose 1-phosphate. Our lab is exploring the idea of using a combination of the purine analogue 2-fluoroadenine (2FA) combined with the MTAP-substrate MTA to specifically target MTAP-deleted tumor cells. The selectivity of this combination relies on three key points: 1) 2FA is actually a prodrug that must first be converted to 2-fluoroadenosine monophosphate (2FAMP) to be active; 2) high concentrations of adenine can inhibit the conversion of 2FA to 2FAMP; and 3) MTA can be converted to adenine only in cells that express MTAP. Thus, MTA will protect normal tissues from 2FA toxicity, while the MTAP-deficient tumor will be left vulnerable. We refer to this approach as the MTA protection strategy. Studies so far in the lab has shown that the combination of 2FA+MTA can selectively kill MTAP- cells in vitro, and that in vivo MTA addition can greatly reduce the toxicity of 2FA in a variety of tissues. In xenograft tumor models in mice, 2FA+MTA slows tumor growth of MTAP- tumors, but the effectiveness of the treatment is not as striking as observed in vivo. Current efforts in the lab are focused toward understanding the mechanistic and pharmacokinetic properties of 2FA+MTA in an effort to develop a more effective regimen. The overall goal of this work is to optimize the use of this drug combination in mice with the ultimate goal of translation into human patients.
Cystathionine beta-synthase (CBS) encodes an enzyme that plays a key role in the transsulfuration pathway that allows the conversion of methionine to cysteine. Loss of function mutations in the CBS gene causes CBS deficiency (or classic homocystinuria), a rare inborn error of metabolism. This condition is characterized by extremely elevated levels of plasma total homocysteine (tHcy), which causes a variety of clinical problems in the vascular, visual, skeletal, and nervous systems. Our lab has developed a variety of humanized mouse models for CBS deficiency in which the mouse gene has been replaced by a human gene with a mutation found in patients with homocystinuria. We are using these models to understand the biology of CBS deficiency, but also as a tool to develop novel treatments. Currently, we are using these models to evaluate a candidate treatment involving rAAV-based gene therapy. Our preliminary data suggests that this technique may be extremely promising to treat the disease.
In the United States, there are over 50,000 cases of Renal Cell Carcinoma (RCC) diagnosed annually resulting in more than 13,000 deaths. Prognosis in RCC is very much dependent on the stage at which the disease is caught. Small tumors confined to the kidney (T1) have 5-year survival rates as high as 90%, while advanced tumors that have metastasized outside the kidney (T4) have rates less than 20%. Unfortunately, most individuals with locally confined disease have no obvious symptoms and, therefore, the disease is often detected at an advanced stage. Increasing the percentage of individuals diagnosed with early stage disease would lead to a significant reduction in mortality from this disease. The development of an inexpensive blood based screening test to detect RCC would be extremely useful in achieving this goal. Our lab is examining the possibility of using blood amino acid levels as potential biomarkers for the early detection of RCC. In retrospective studies we have consistently observed that the serum amino acid profile could be used to distinguish RCC patients from controls with excellent specificity and sensitivity. We are now trying to determine whether this difference holds in a prospective cohort. To this end we have set up a collaboration with the Janus serum bank in Norway. This bank has over 318,000 serum samples that are linked prospectively to 95,000 cancer patients. By utilizing this rescource we will be able to determine the utility of amino acid profiling in RCC detection.
This Fox Chase professor participates in the Undergraduate Summer Research Fellowship.
Learn more about Research Volunteering.




