Israel Cañadas, PhD

Assistant Professor
Member, Cancer Epigenetics Institute
Research Program
Lab Overview
Co-opting endogenous retroviral signaling as a lung cancer vulnerability
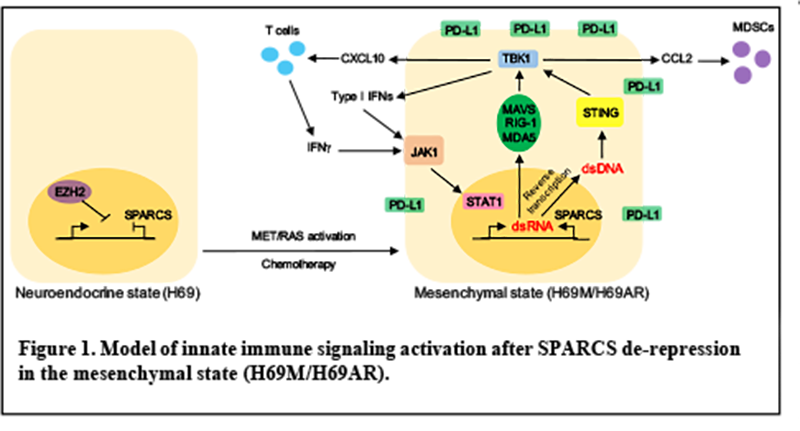
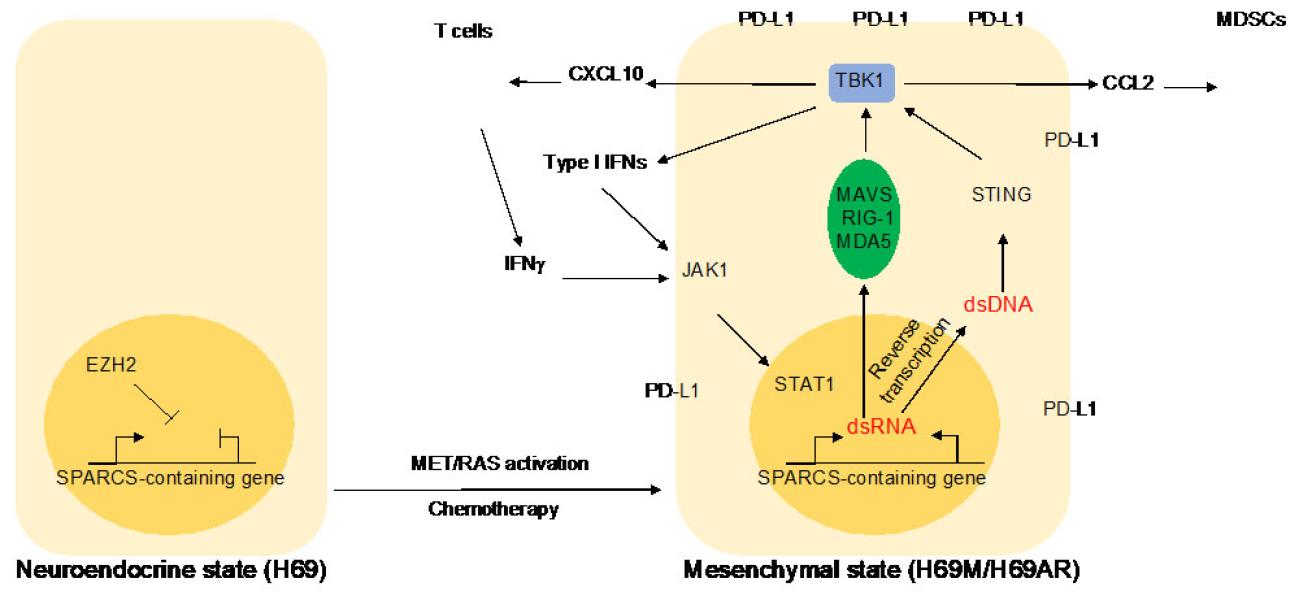
We recently identified a novel epigenetically regulated subclass of endogenous retroviruses (ERVs) that engages pathologic innate immune signaling in mesenchymal cancer subpopulations of Small Cell Lung Cancer (SCLC), with potentially important implications for cancer immunotherapy (Cañadas et al, Nat Med, 2018). Stimulated 3 Prime Antisense Retroviral Coding Sequences (SPARCS) are oriented inversely in 3’UTRs of certain interferon-inducible genes and silenced by EZH2. De-repression of these loci resulted in dsRNA generation following IFNs exposure due to bi-directional transcription from the STAT1-activated gene promoter and the 5’ LTR of the antisense ERV.
The overarching goal of this project is to study the biological mechanisms that connect the mesenchymal cancer resistant state to SPARCS de-repression in lung cancer, the impact on the tumor microenvironment, and co-opting this cell state to favor response to immune checkpoint blockade.
Ex vivo organotypic cultures
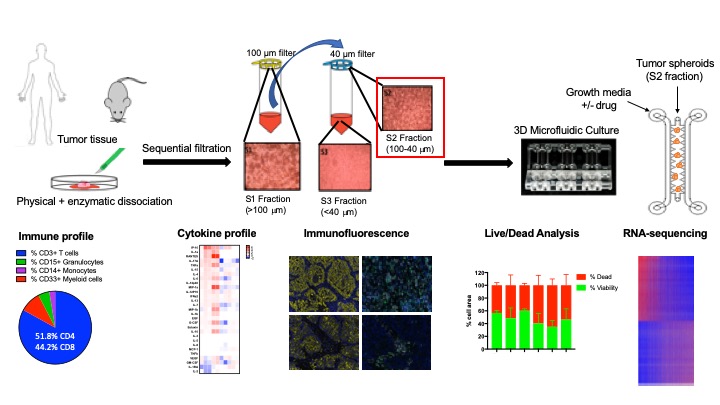
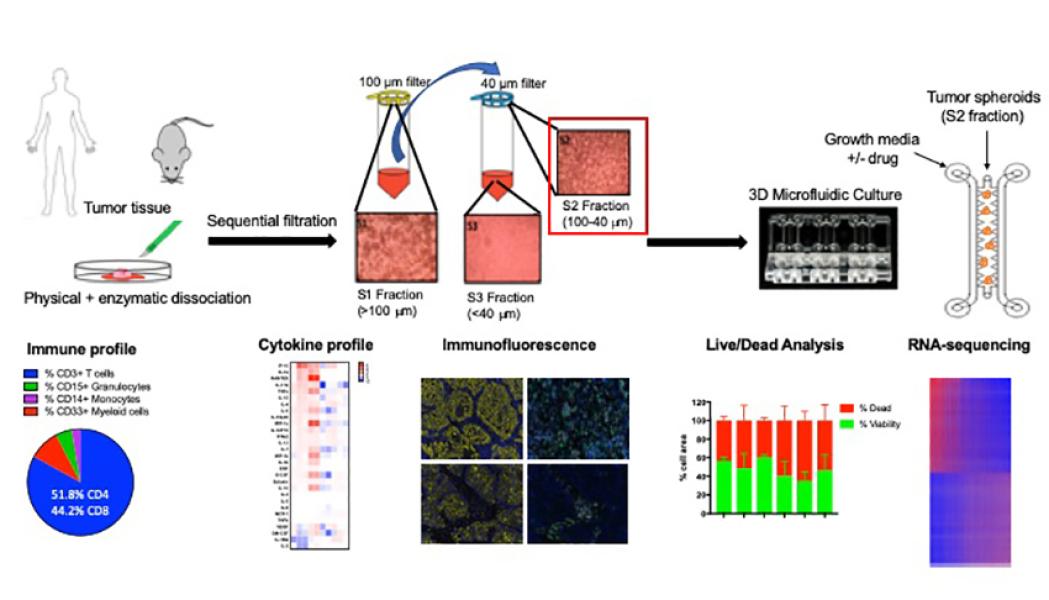
We are developing novel functional ex vivo organotypic 3D culture platforms using surgically resected murine and patient-derived tumor samples that will allow us to validate promising therapeutic combinations in an ex vivo system that incorporates features of the tumor microenvironment and models the dynamic response to immune checkpoint blockade (Jenkins et al, Cancer Discovery, 2018). Using this sophisticated approach, we aim to identify and characterize novel mechanisms of response and resistance to immunotherapy.
Small Cell Lung Cancer
Small cell lung cancer (SCLC) is an aggressive and lethal lung malignancy with a 5-year overall survival of less than 5%. While having one of the highest mutational burdens because of its strong association with tobacco smoking, SCLC is often characterized by a reduced antigen presentation and an immunosuppressive tumor microenvironment. Therefore, despite promising advances in the use of immunotherapy, only a fraction of SCLC patients respond to these therapies.
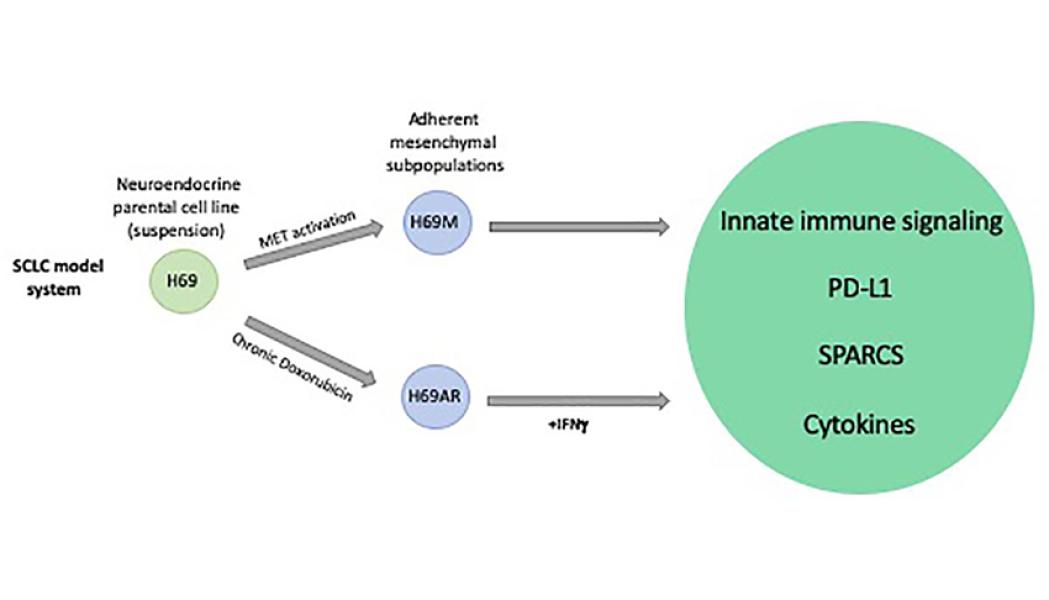
SCLC is composed of phenotypically different cells with either neuroendocrine or mesenchymal features. Of note, the mesenchymal compartment enhances chemoresistance and metastatic potential of neuroendocrine cells. We and others have shown that activation of MET and/or RAS signaling can fuel this mesenchymal phenotype, resulting in chemoresistance and increased tumorigenesis in a well-established SCLC model (Cañadas et al, Clin Cancer Res, 2014). In addition, we recently showed an enhanced innate immune signaling in these SCLC mesenchymal subpopulations because of the de-repression of a novel epigenetically regulated subclass of ERVs (Cañadas et al, Nat Med, 2018).
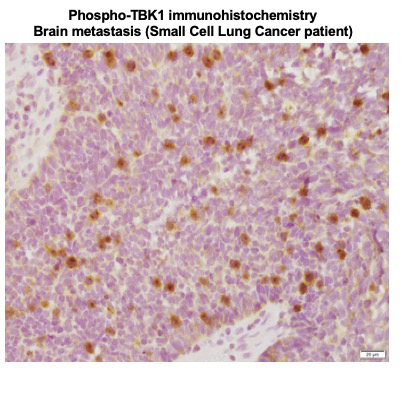
Using SCLC as a model, we aim to identify and characterize biological mechanisms by which intratumor heterogeneity may influence the tumor microenvironment and response to therapy, providing insights in tumor immunology and informing clinical strategies to improve immunotherapies in SCLC.
Educational Background
- PhD, Biomedicine, Pompeu Fabra University (UPF), Barcelona, Spain, 2013
- MS, Biomedical Research, Pompeu Fabra University (UPF), Barcelona, Spain, 2009
- BS, Biology, Barcelona University (UB), Barcelona, Spain, 2007
Honors & Awards
- The G. Harold and Leila Y. Mathers Foundation Award (2020)
- W. W. Smith Charitable Trust Grant (2020)
- 2018 Lung Cancer Research Foundation Annual Grant Recipient
- Short-Term fellowship - Red Temática de Investigación Cooperativa en Cáncer (RTICC) (2014)
- Summa cum laude distinction of Doctoral Thesis, 2013
- PhD fellowship - Institut Hospital del Mar d’Investigacions Mèdiques (IMIM) (2009)
People



Research Interests
Cañadas Lab: Targeting innate immunity in lung cancer
- Study of tumor immunity and the tumor microenvironment in lung cancer
- Deciphering mechanisms of Endogenous Retroviruses (ERV) silencing and reactivation at the epigenetic level.
- Study of cytosolic nucleic acid-sensing pathways in the context of innate and adaptive antitumor immunity.
- Development of novel functional ex vivo organotypic 3D culture platforms to identify and characterize mechanisms of response and resistance to immunotherapy using murine and patient derived tumor samples.
Lab Overview
The Cañadas Lab studies tumor immunity and the tumor microenvironment in lung cancer with the ultimate goal of identifying new therapeutic opportunities to improve patient health.
We are primarily interested in delineating mechanisms of tumor resistance to immunotherapy, and in leveraging innate immune signaling pathways to break therapy resistance and restore immunogenicity.
Selected Publications
Campisi M, Sundararaman SK, Shelton SE, Knelson EH, Mahadevan NR, Yoshida R, Tani T, Ivanova E, Cañadas I, Osaki T, Ling Lee SW, Thai T, Han S, Piel BP, Gilhooley S, Paweletz CP, Chiono V, Kamm RD, Kitajima S, Barbie DA. Tumor-Derived cGAMP Regulates Activation of the Vasculature. Front. Immunol. 2020 Sep 4;11:2090. doi: 10.3389/fimmu.2020.02090.
Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su MJ, Melms JC, Leeson R, Kanodia A, Mei S, Lin JR, Wang S, Rabasha B, Liu D, Zhang G, Margolais C, Ashenberg O, Ott PA, Buchbinder EI, Haq R, Hodi FS, Boland GM, Sullivan RJ, Frederick DT, Miao B, Moll T, Flaherty KT, Herlyn M, Jenkins RW, Thummalapalli R, Kowalczyk MS, Cañadas I, Schilling B, Cartwright ANR, Luoma AM, Malu S, Hwu P, Bernatchez C, Forget MA, Barbie DA, Shalek AK, Tirosh I, Sorger PK, Wucherpfennig K, Van Allen EM, Schadendorf D, Johnson BE, Rotem A, Rozenblatt-Rosen O, Garraway LA, Yoon CH, Izar B, Regev A. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell. 2018 Nov 1;175(4):984-997.e24. doi: 10.1016/j.cell.2018.09.006.
Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, Mathur N, Zhou C, Coakley RV, Bartels A, Bowden M, Herbert Z, Hill S, Gilhooley S, Carter J, Cañadas I, Thai TC, Kitajima S, Chiono V, Paweletz CP, Barbie DA, Kamm RD, Jenkins RW. 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab Chip. 2018 Sep 5. doi: 10.1039/c8lc00322j.
Cañadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, Campisi C, Kuang Y, Zhang Y, Gjini E, Zhang G, Tian T, Sen DR, Miao D, Imamura Y, Thai T, Piel B, Terai H, Aref AR, Hagan T, Koyama S, Watanabe M, Baba H, Adeni AE, Lydon CA, Tamayo P, Wei Z, Herlyn M, Barbie TU, Uppaluri R, Sholl LM, Sicinska E, Sands J, Rodig S, Wong KK, Paweletz CP, Watanabe H, Barbie DA. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med. 2018 Aug;24(8):1143-1150.
Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, He MX, Walker W, Zhang G, Tian T, Cheng C, Wei Z, Palakurthi S, Bittinger M, Vitzthum H, Kim JW, Merlino A, Quinn M, Venkataramani C, Kaplan JA, Portell A, Gokhale PC, Phillips B, Smart A, Rotem A, Jones RE, Keogh L, Anguiano M, Stapleton L, Jia Z, Barzily-Rokni M, Cañadas I, Thai TC, Hammond MR, Vlahos R, Wang ES, Zhang H, Li S, Hanna GJ, Huang W, Hoang MP, Piris A, Eliane JP, Stemmer-Rachamimov AO, Cameron L, Su MJ, Shah P, Izar B, Thakuria M, LeBoeuf NR, Rabinowits G, Gunda V, Parangi S, Cleary JM, Miller BC, Kitajima S, Thummalapalli R, Miao B, Barbie TU, Sivathanu V, Wong J, Richards WG, Bueno R, Yoon CH, Miret J, Herlyn M, Garraway LA,
Van Allen EM, Freeman GJ, Kirschmeier PT, Lorch JH, Ott PA, Hodi FS, Flaherty KT, Kamm RD, Boland GM, Wong KK, Dornan D, Paweletz CP, Barbie DA. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov. 2018 Feb;8(2):196-215.
Jenkins RW, Thummalapalli R, Carter J, Cañadas I, Barbie DA. Molecular and Genomic Determinants of Response to Immune Checkpoint Inhibition in Cancer. Annu Rev Med. 2018 Jan 29;69:333-347.
Yang S, Imamura Y, Jenkins RW, Cañadas I, Kitajima S, Aref A, Brannon A, Oki E, Castoreno A, Zhu Z, Thai T, Reibel J, Qian Z, Ogino S, Wong KK, Baba H, Kimmelman AC, Pasca Di Magliano M, Barbie DA. Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation. Cancer Immunol Res. 2016 Jun;4(6):520-30.
Sánchez-Martín FJ, Bellosillo B, Gelabert M, Dalmases A, Cañadas I, Vidal J, Martinez A, Argilés G, Siravegna G, Arena S, Koefoed K, Visa L, Arpí O, Horak ID, Iglesias M, Stroh C, Kragh M, Rovira A, Albanell J, Tabernero J, Bardelli A, Montagut C. The first-in-class anti-EGFR antibody mixture Sym004 overcomes cetuximab-resistance mediated by EGFR extracellular domain mutations in colorectal cancer. Clin Cancer Res 2016 Jul 1;22(13):3260-7.
Cañadas I, Taus A, Villanueva X, Arpí O, Pijuan L, Rodríguez Y, Menéndez S, Mojal S, Rojo F, Albanell J, Rovira A, Arriola E. Angiopoietin-2 is a negative prognostic marker in small cell lung cancer. Lung Cancer 2015 Nov;90(2):302-6.
Casadevall D, Gimeno J, Clavé S, Taus A, Pijuan L, Arumí M, Lorenzo M, Menéndez S, Cañadas I, Albanell J, Serrano S, Espinet B, Salido M, Arriola E. MET expression and copy number heterogeneity in nonsquamous non-small cell lung cancer (nsNSCLC). Oncotarget 2015; 30;6(18):16215-26.
Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, Lazzari L, Ferruz N, Russo M, Misale S, González I, Iglesias M, Gavilan E, Corti G, Hobor S, Salido M, Sánchez J, Dalmases A, Bellmunt J, De Fabritiis G, Rovira A, Di Nicolantonio F, Albanell J, Bardelli A, Montagut C. Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res 2015; 21(9):2157-66
Cañadas I, Taus A, González I, Villanueva X, Gimeno J, Pijuan L, Domine M, Sánchez-Font A, Vollmer I, Menéndez S, Arpí O, Mojal S, Rojo F, Rovira A, Albanell J, Arriola E. High circulating hepatocyte growth factor levels associate with epithelial to mesenchymal transition and poor outcome in small cell lung cancer patients. Oncotarget 2014; 5(14): 5246-5256.
Cañadas I, Rojo F, Taus A, Arpí O, Arumí M, Pijuan L, Menéndez S, Zazo S, Domine M, Salido M, Mojal S, García de Herreros A, Rovira A, Albanell J, Arriola E. Targeting epithelial to mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin Cancer Res 2014; 20(4): 938-950.
Salido M, Pijuan L, Galvan AB, Gimeno J, Cañadas I, Rodríguez M, Rojo F, Albanell J, Solé F, Arriola E. ALK status in a primary lung tumour and metachronous metastases. Histopathology 2012; 60(5): 843-845.
Arriola E*, Cañadas I*, Arumí M, Domine M, López-Vilariño JA, Arpí O, Salido M, Menéndez S, Grande E, Hirsch FR, Serrano S, Bellosillo B, Rojo F, Rovira A, Albanell J. MET phosphorylation predicts poor outcome in small cell lung carcinoma and its inhibition blocks HGF-induced effects in MET mutant cell lines. Br J Cancer 2011; 105(6): 814-823.
*equal contribution.
Salido M, Pijuan L, Martínez-Avilés L, Galvan AB, Cañadas I, Rovira A, Zanui M, Martínez A, Longarón R, Solé F, Serrano S, Bellosillo B, Wynes MW, Albanell J, Hirsch FR, Arriola E. Increased ALK Gene Copy Number and Amplification are Frequent in Non-small Cell Lung Cancer. J Thorac Oncol 2011; 6(1): 21-27.




